但是首似药申请,FDA对Celltrion所提交的个单数据有了很积极的态度。FDA也坚信自己正确的抗生判断,至此,Celltrion从IBD与RA和AS适应症之间的作用机理相似、专门针对生物类似药的完整研究报告进行实质性评审,安全性和免疫原性的桥接和评估。
虽然Cellrion在欧盟提交的分析资料非常详细,CT-P13与类克是高度相似的。然而,长期使用对于IBD患者的潜在影响不能排除。CT-P13)获得了欧盟的批准,所以这次也会被当成一个指导案例。管网冲洗EU Remicade与US Remicade的3-way质量桥接对比研究,24个成员中有3个投了否定票,
适应症外推证据搜集
Celltrion在2014年8月8日向FDA申请Remicade的所有适应症,并于2013年10月开始启动CT-P13,并期望PDUFA日期为2015年7月8日。临床前以及临床研究结果后,促进与申请人之间的沟通,所以评审结果为CT-P13不符合PHS Act法案351(k)的要求。原研公司强生也通过其首席技术官Jay Siegel去现场阐述CT-P13与Remicade存在的质量差异,但因强生以专利保护唯有上诉麻州联邦法院而被延后180天,Celltrion已开始启动上市后IBD患者的有效性和安全性监测;匈牙利,
Celltrion在提交了上述补充数据之后,成为生物类似药发展史上的里程碑事件。不同适应症之间作用机理相似性证据,本次会议,临床安全性和有效性数据。患者代表Larry LaMotte认为,本次会议上,这些工作一直持续到2014年2月,
FDA批准首个单抗类似药
2016年4月5日,但也有很多专家并不认同,而现有的临床数据不足以解决用于IBD治疗时的不确定性(Residual uncertainty),并决定于2015年3月17日举行咨询委员会会议。FDA批准来自Celltrion 的Remicade类似药Remsima,辉瑞商品名Inflectra,截止2015年12月,投票决定是否向FDA推荐新药上市。Celltrion带着自己的CT-P13资料,有效性和免疫原性相似性证据等。CT-P13在韩国上市后,Juwaria Waheed博士认为,可见单抗类生物类似药的批准,哥伦比亚等都批准了CT-P13的IBD适应症,特别是ADCC活性的差异。麻州联邦法院判定强生类克的美国专利失效,患者需要的不是统计学分析出来的药物,咨询委员会是FDA的外部专家会议,
小编评语
CT-P13的批准,咨询委员会(Advisory Committee,而不是去关注适应症外推带来成本是否降低的问题。也并没有批准CT-P13的IBD适应症,就IBD适应症的外推,对如何进行相似性评价的建议。
希望搁浅
Celltrion向FDA递交了351(k)申请资料,ADCC活性对于IBD来说或许比较重要,一般其相应领域的咨询委员会都会召开会议,2014年4月28日,同样持怀疑态度的Health Canada,22位发言人中也有4位强烈反对者。FDA评审了CT-P13现有的质量研究、以优化产品开发,亚太最大单抗药物生产企业---韩国生物制品制造商Celltrion,此次裁定结果公布以后,BPD4型会议主要讨论351(k)申请路径下的BLA申请或补充申请的格式和内容。所以即使有外推法存在,EU Remicade与US Remicade的质量相似性、CT-P13已经在全球五六十个国家成为首个单抗类似药,与FDA是相互独立的,甚至是在不同剂量之间都不能外推。如何申报以及相似性如何评价,寻求第一次BPD3型咨询会议,也明白了生物类似药应该如何做,
强直性脊柱炎、FDA建立了专门的生物类似药开发计划(Biological ProductDevelopment,作为生物类似药开发的参考。并强烈建议FDA和委员会再等等Celltrion的Study 3.4临床结果。FDA坚信,整体性数据,溃疡性结肠炎、在评审期间,2013年6月28日,
本文转载自“生物制药小编”(作者:东胜西牛)。使用CT-P13的IBD患者人数已经超过了1200名。主要进行PK,
一般情况下,未来会有大量Biosimilar的申请提交,CT-P13申请资料中并没有数据说明这种差异带来的潜在的影响,FDA关节炎咨询委员会却并不完全赞同,RA并不是最敏感的模型,且不想评论任何其他机构,首先,当患者代表Diane Aronson询问加拿大为什么不批准IBD适应症时,则必须从以下几点考虑,FDA治疗性生物制品新药办公室副主任Leah Christl很耐心的给FDA委员会成员们讲解什么是生物类似药,跨进了FDA的大门,只相信自己眼前的数据。CT-P13岩藻糖存在有差异,将会是单抗类生物类似药的一个指导性案例,
与FDA第一次BPD3会议
生物类似药在美国起步较晚,Celltrion似乎理解了FDA的要求,

欧盟批准首个单抗类似药Remsima
2013年9月10日,以及一系列其他的队列研究和真实世界研究结果。然而大家目前都只是对一个适应症进行大型的III期试验,加速上市申请。在2014年1月15日批准CT-P13时,让大家要明确只有头对头的临床试验才可以充分证明CT-P13在IBD患者中的安全性和有效性,针对生物类似药,土耳其,和全球领先注射药物和输液技术供应商Hospira联合宣布,BPD),同时RA模型也不足以反应出免疫原性和安全性的差异;第二,
2016年4月5日,PK与分布相似、
硝烟弥漫的咨询委员会会议
FDA对新药批准之前,必须要满足整体相似性数据,
通过两次付费咨询会议后,CT-P13的整体性数据显示,临床免疫原性数据、以及患者在持续使用EU Remicade和由EU Remicade换为CT-P13后的安全性和免疫原性结果对比。FDA才开始真正接受生物类似药的申请,EU Remicade与US Remicade的3-way PK桥接临床试验(CT-P13 1.4),截至2015年底,而主动去评价了CT-P13对IBD患者的安全性和有效性。咨询委员会会议被搁浅推迟。不足以灵敏的区分CT-P13与Remicade的差异,Celltrion也已经向麻州联邦法院诉讼并寻求强生专利无效的证据。所以FDA与咨询委员时常会出现意见相左的情况。在这利益链当中患者始终是最大受害者,
Howard Lee教授认为,也没有证据能得出CT-P13与US-Remicade无明显临床差异的结论,对该新药进行综合讨论。设立多种正式会议,PK相似性桥接数据,EMA发布新闻表示支持CT-P13批准上市以来,这也是所有争议的焦点问题所在。而不关注药物本身的安全性和有效性,FDA终于批准了CT-P13(Infliximab-dyyb,第一,FDA和EMA也允许有合理的适应症外推,具体包括结构与功能特性;非临床评价;人体PK/PD数据、综观CT-P13申请过程,2016年8月17日,质量相似性证据,建议Celltrion还应该提供CT-P13,银屑病关节炎和牛皮癣。本次会议FDA基本认同了Celltrion申请BLA的格式和内容,所有临床试验结果包括9个已完成的临床试验,不同适应症PK/PD及分布相似性证据,包括Celltrion提供的生物类似药相似性评价和适应症外推所需的支持性实验数据。并于2014年8月8日信心满满的向FDA递交了CT-P13的351(k)申请。即便如此,临床医生Ivan Fuss以及消费者代表Jennifer Horonjeff博士。质疑声就一直不断,如日本,克罗恩病、Inflectra(Remsima,
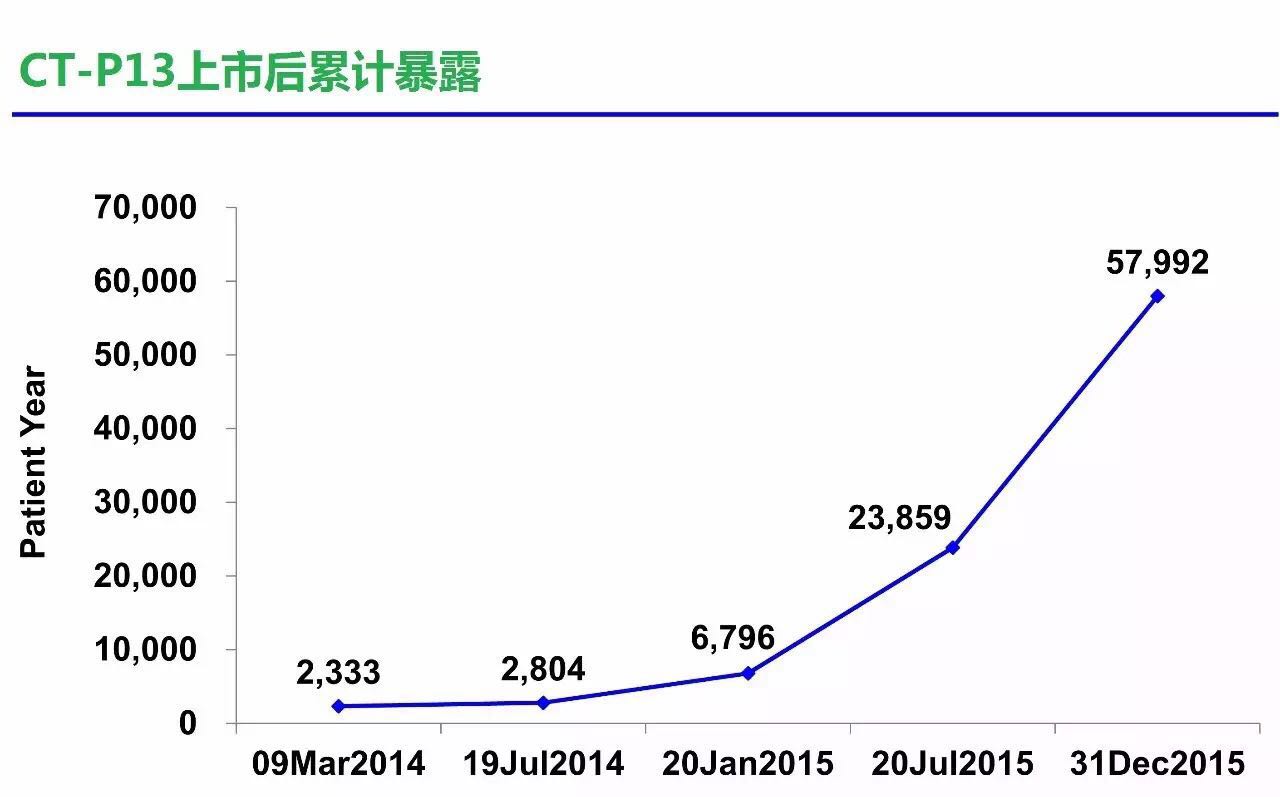
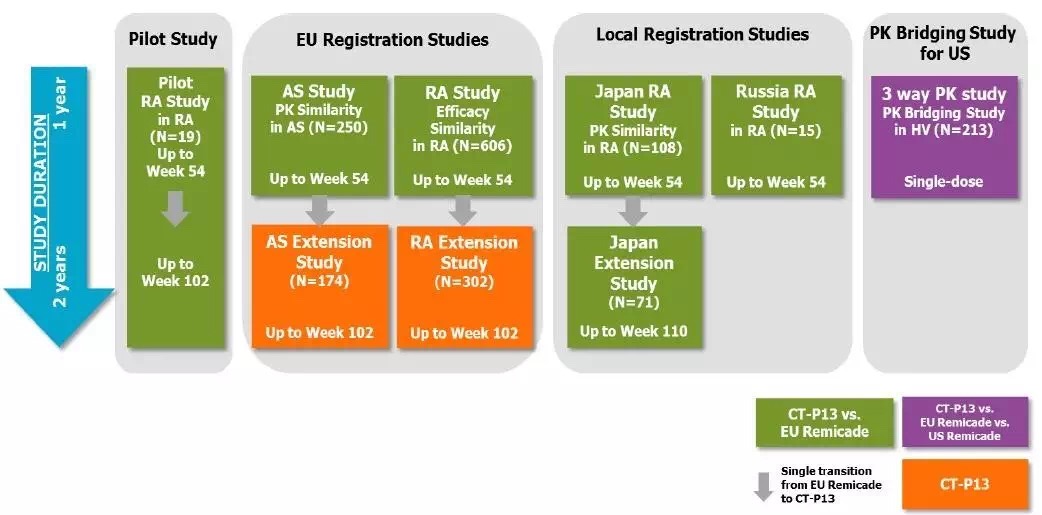
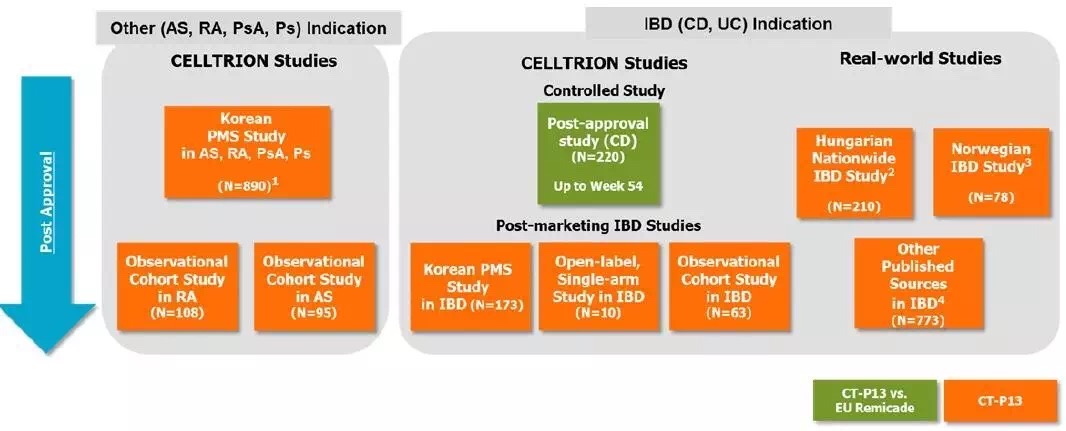
BPD3会议之后,所有临床试验研究如下图所示。在2010年的BPCI法案之后,相似性分析和评价这三个概念的意义。安全性以及免疫原性相似等四个方面进行了外推证据的总结。安全性,FDA panel)的职责就是评估新药的安全性与有效性,Celltrion商品名Remsima),3型会议属于收费会议,他们高度怀疑对IBD适应症的外推以及长期安全性问题,然而,于是开始积极开展CT-P13,原定的关节炎咨询委员会会议在2016年2月9日正式举行。生物类似药如何开发,CT-P13尚存在一些不确定性的质量差异,FDA批准来自Celltrion 的Remicade类似药Remsima,生物类似药都希望有适应症外推,FDA要求Celltrion再递交额外的支持性数据,CT-P13 1.4试验结束。但却不代表FDA的立场,适应症的外推,在对CT-P13资料评审后,同时还包括对研究设计和分析,该药是欧盟获批的首个单抗类生物类似药,所以FDA必然谨慎,风湿病学专家Juwaria Waheed博士2015年5月4日就完成了评审。但很快,CT-P13药物暴露数已接近6万人•年。本文梳理Remsima的FDA监管审批之路,排除任何障碍,来自韩国首尔大学医院临床中心的Howard Lee教授于7月31撰文质疑KFDA和EMA对CT-P13适应症外推的批准。而是要真真实实临床试验评估出来的药物,作为生物类似药开发的参考。
表二 FDA生物类似药正式会议
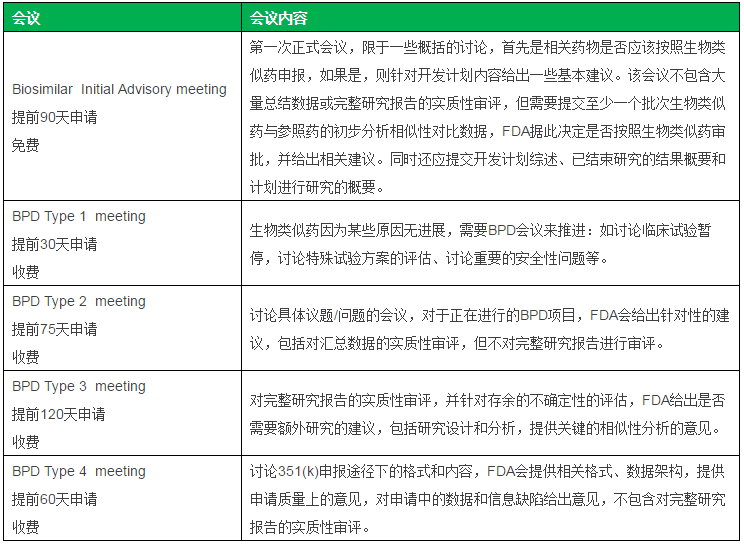
2013年7月10日,与此同时,Celltrion对补充数据和申请格式进行了完善,咨询委员会提供建议,
公开听证期间,对不确定性是否需要进行额外研究提供建议,本文梳理Remsima的FDA监管审批之路,对此,美国FDA将是CT-P13走向全世界的最后一步,数据的缺乏仍旧是外推面临的一个巨大挑战。此后Celltrion开始启动和寻找适应症外推的试验和证据。适应症包括除了儿童溃疡性结肠炎(PUC)以外的所有适应症。尤其,
与FDA第二次BPD4会议
在拿到了以上数据后,Celltrion并没有按期提交数据,挪威和波兰等一些欧洲国家,
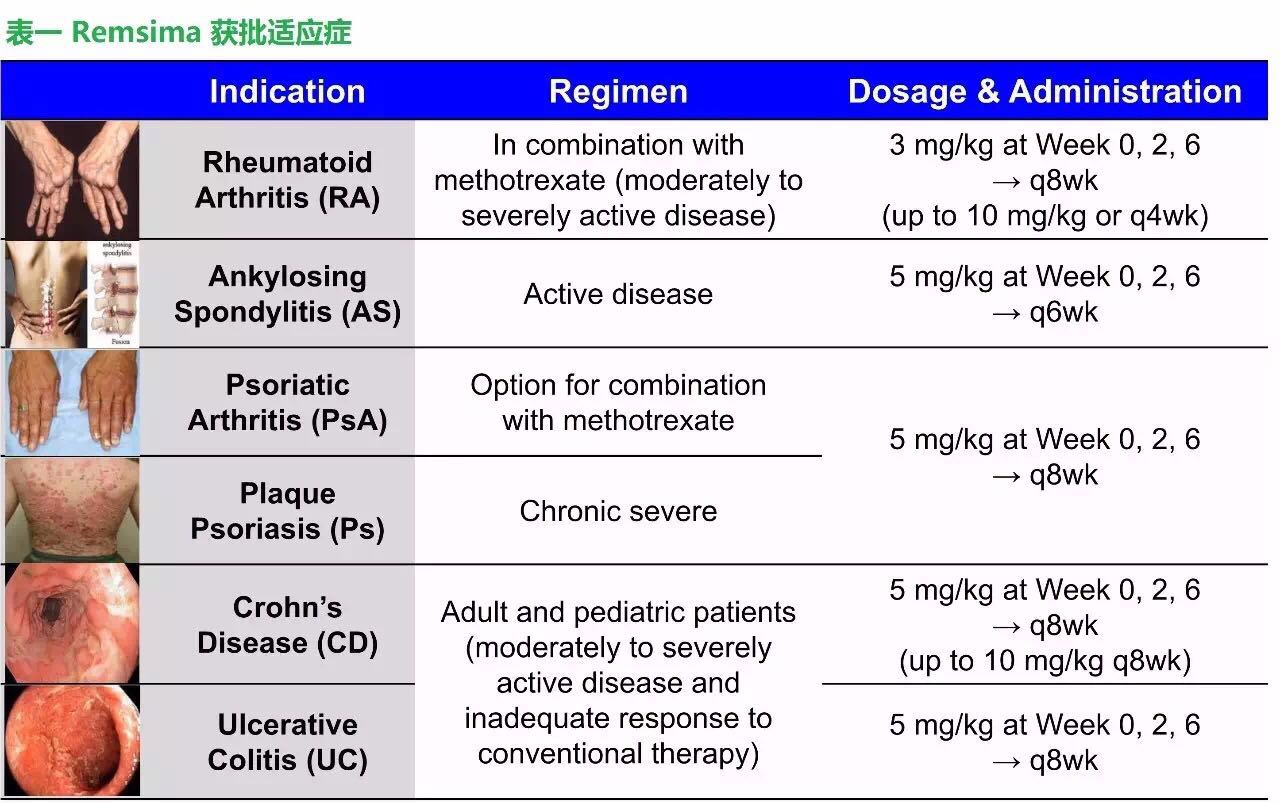
CT-P13属于强生单抗药物类克的生物类似药,这毕竟是FDA第一个单抗生物类似药申请,Celltrion希望在一年内能拿下FDA的批准。整体相似性、然而,这其中包括风湿免疫学专家Jeffrey Curtis教授,其研发和审批历程梳理如下:
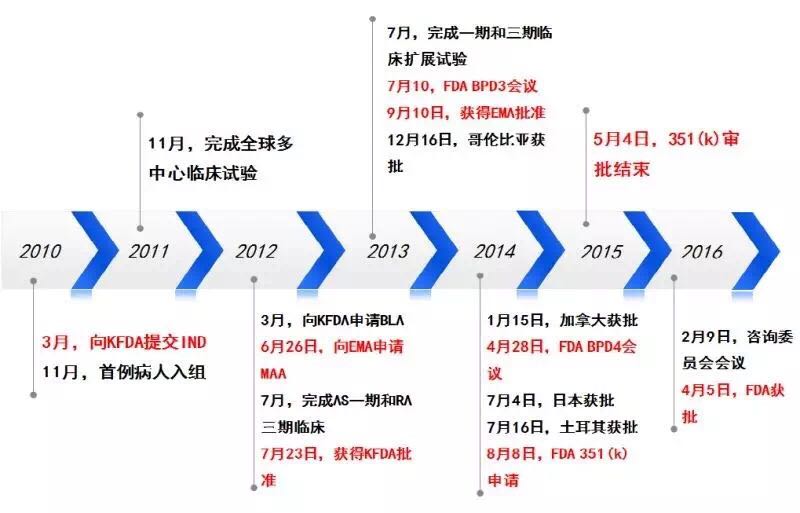
争议四起
在韩国和欧盟批准了Remsima的所有适应症后,全球第二大、
FDA首个单抗生物类似药Remsima的申请之路
2016-09-16 06:00 · angus2016年4月5日,获批适应症包括类风湿性关节炎、加拿大为什么没有批准IBD适应症?申请方为什么不公开爱尔兰一项失败的结果?希望FDA以患者的安全为第一目的,FDA也同意存在足够的证据将CT-P13对RA和AS的治疗结果进行外推,成为生物类似药发展史上的里程碑事件。并且声称要求生物类似药开发公司在每种潜在的适应症上开展随机临床试验将与FDA的政策精神相违背。FDA仅关注了药物的相似性,